Авторизация
Поиск по указателям
Leach A.R. — Molecular Modelling Principles and Applications
Обсудите книгу на научном форуме Нашли опечатку?
Название: Molecular Modelling Principles and ApplicationsАвтор: Leach A.R. Аннотация: Preface to the Second Edition The impetus for this second edition is a desire to include some of the new techniques that have emerged in recent years and also extend the scope of the book to cover certain areas that were under-represented (even neglected) in the first edition. In this second volume there are three topics that fall into the first category (density functional theory, bioinformatics/protein structure analysis and chemoinformatics) and one main area in the second category (modelling of the solid state). In addition, of course, a new edition provides an opportunity to take a critical view of the text and to re-organise and update the material. Thus whilst much remains from the first edition, and this second book follows much the same path through the subject, readers familiar with the first edition will find some changes which I hope they will agree are for the better. As with the first edition we initially consider quantum mechanics, but this is now split into two chapters. Thus Chapter 2 provides an introduction to the ab initio and semi-empirical approaches together with some examples of the uses of quantum mechanics. Chapter 3 covers more advanced aspects of the ab initio approach, density functional theory and the particular problems of the solid state. Molecular mechanics is the subject of Chapter 4 and then in Chapter 5 we consider energy minimisation and other 'static' techniques. Chapters 6, 7 and 8 deal with the two main simulation methods (molecular dynamics and Monte Carlo). Chapter 9 is devoted to the conformational analysis of 'small' molecules but also includes some topics (e.g. cluster analysis, principal components analysis) that are widely used in informatics. In Chapter 10 the problems of protein structure prediction and protein folding are considered; this chapter also contains an introduction to some of the more widely used methods in bioinformatics. In Chapter 11 we draw upon material from the previous chapters in a discussion of free energy calculations, continuum solvent models, and methods for simulating chemical reactions and defects in solids. Finally, Chapter 12 is concerned with modelling and chemoinformatics techniques for discovering and designing new molecules, including database searching, docking, de novo design, quantitative structure-activity relationships and combinatorial library design. As in the first edition, the inexorable pace of change means that what is currently considered 'cutting edge' will soon become routine. The examples are thus chosen primarily because they illuminate the underlying theory rather than because they are the first application of a particular technique or are the most recent available. In a similar vein, it is impossible in a volume such as this to even attempt to cover everything and so there are undoubtedly areas which are under-represented. This is not intended to be a definitive historical account or a review of the current state-of-the-art. Thus, whilst I have tried to include many literature references it is possible that the invention of some technique may appear to be incorrectly attributed or a 'classic' application may be missing. A general guiding principle has been to focus on those techniques that are in widespread use rather than those which are the province of one particular research group. Despite these caveats I hope that the coverage is sufficient to provide a solid introduction to the main areas and also that those readers who are 'experts' will find something new to interest them.
Язык: Рубрика: Химия /Статус предметного указателя: Готов указатель с номерами страниц ed2k: ed2k stats Издание: 2-ndГод издания: 2001Количество страниц: 774Добавлена в каталог: 21.02.2007Операции: Положить на полку |
Скопировать ссылку для форума | Скопировать ID
Предметный указатель
Force field models, empirical, angle bending 166 173 Force field models, empirical, bond stretching 166 170—173 Force field models, empirical, Class 1, 2 and 3 178—180 Force field models, empirical, cross terms 178—180 Force field models, empirical, derivatives of molecular mechanics energy function 225—226 Force field models, empirical, Drude molecules, interaction between 246—247 Force field models, empirical, effective pair potentials 214—215 Force field models, empirical, general features 168—170 Force field models, empirical, hydrogen bonding 215—216 Force field models, empirical, improper torsions and out-of-plane bending 176—178 Force field models, empirical, inorganic molecules 234—236 Force field models, empirical, many-body effects in empirical potentials 212—214 Force field models, empirical, metals and semiconductors 240—245 Force field models, empirical, parameters 221 224—225 228—232 Force field models, empirical, simple 165—166 Force field models, empirical, solid-state systems 236—240 Force field models, empirical, thermodynamic properties calculated using 226—228 Force field models, empirical, torsional terms 173—176 Force field models, empirical, united atom, reduced representations and 221—225 Force field models, empirical, water simulation 216—220 (see also “Non-bonded interactions”) Force-bias Monte Carlo method 432—433 Formaldehyde 76—77 236 Formamide, electron density around 78—79 81—82 Formamide, gradient vector path 81 Formamide, HOMO and LUMO for 79 Forward sampling 567 Fourier, analysis 379 392—393 Fourier, coefficient 148—149 Fourier, series 21—23 155 235 237 392 Fourier, transform 22—24 392 Fractional factorial design 697 Fragments 472 Fragments, binding 689—691 Fragments, conformational analysis 464—465 472 Fragments, locating 687—689 Fragments, marking 715—717 Free energy calculations 563—639 Free energy calculations, chemical reactions 610—622 Free energy calculations, computer difficulties 563—564 Free energy calculations, enthalpy and entropy differences 574 Free energy calculations, linear response method 631—632 Free energy calculations, partitioning 574—576 Free energy calculations, pitfalls 577—570 Free energy calculations, potentials of mean force 580—585 Free energy calculations, rapid methods, approximate 585—592 Free energy calculations, solid-state defects 622—630 (see also “Differences free Helmholtz “Solvation”) Freely rotating chain model 428—429 Frenkel defect 623 626 Friction coefficient 388—389 Frontier orbitals 293 Full configuration interaction 112 Full factorial design 697 Fullerenes 101 Functional genomics 512 future 160—161 718—719 G3 theory 116—117 GA see “Genetic algorithms” Gap penalties 526—528 Gasteiger — Marsili approach 192—193 Gaussian functions/distribution 20—21 Gaussian functions/distribution, basis sets 65—73 passim 120 137 195 Gaussian functions/distribution, computer simulation 336—337 339 Gaussian functions/distribution, conformational analysis 481—482 Gaussian functions/distribution, density functional theory 131—132 Gaussian functions/distribution, force fields 195—196 Gaussian functions/distribution, Gaussian-3 (G3) theory 116—117 Gaussian functions/distribution, many-body perturbations 116—117 Gaussian functions/distribution, molecular dynamics simulation 365 381 384 389—390 Gaussian functions/distribution, new molecules 679 703 Gaussian functions/distribution, proteins 551 Gaussian functions/distribution, SCF 119 Gaussian functions/distribution, semi-empirical methods 92—98 Gaussian functions/distribution, solid state quantum mechanics 146 Gay — Berne potential 222—225 GB (generalised Born equation) 598—601 GB (generalised Born equation), surface area model (GB/SA) 609 Gear algorithm 358—359 General polyelectronic systems 38—41 46—50 Generalised coordination 370 Generalised valence bond 125 Generating Optimal Linear PLS Estimations 711 Generator matrices 429 Genetic algorithms, conformational analysis 479—482 Genetic algorithms, new molecules 653 663 691 701 Genomics 512 548—549 Geometry 658—659 Geometry, distance 467—475 476 651—653 663 Germanium 159—160 244 Gibbs ensemble Monte Carlo method 439 450—451 Gibbs free energy 563 569 Global energy minimum 253 458 479—483 551—552 Glutamic acid 510 525 556—557 Glutamine 510 525 556—557 Glycine 221 459 511 525 556—557 Go-Scheraga chain closure algorithm 541—542 Goal nodes 461 GOLD program 667 GOLPE (Generating Optimal Linear PLS Estimations) 711 Gradient, corrected functional 134—135 Gradient, exchange 135 136 137 Gradient, vector path 80—81 Grand canonical Monte Carlo simulations 440—442 Graphics, molecular 5—6 Graphite, adsorption 441—442 graphs 642—643 654 Green — Kubo formula 382 GRID program 215 687—688 708 711 Grid search 459 505 GROMOS program 330 Grotthuss mechanism 620 Group 14 elements 244 Group 14 elements, solid state quantum mechanics applied to 158—60 (see also “Carbon” “Germanium” “Silicon”) Group average 493—494 Group-based cut-offs 327—330 Guanine 227 GVB (generalised valence bond) 125 Haemagglutinin 667 Haemerythrin 544 Halides 237 239 504 571 Halogenated hydrocarbons 707 Hamiltonian operator, advanced ab initio methods 114—115 120—121 Hamiltonian operator, computational quantum mechanics 27—29 30 32 36 42 46 53 90—92 Hamiltonian operator, computer simulation 312 313 410 Hamiltonian operator, force fields 246 Hamiltonian operator, free energy calculations 565 567—569 574 577—579 586 595—596 614 Hammett substituent parameter 695—697 Hamming (city block) distance 492 676—678 Hard-sphere model 353—354 Harmonic approximation 278 Harmonic potential see “Hooke’s law” Hartree atomic unit 29 Hartree product 38—39 Hartree — Fock equations/theory 51—65 85 Hartree — Fock equations/theory, application 65 Hartree — Fock equations/theory, closed-shell 109 Hartree — Fock equations/theory, configuration interaction 112—113 Hartree — Fock equations/theory, density functional theory 126 128 129 135—137 Hartree — Fock equations/theory, free energy calculations 615—616 Hartree — Fock equations/theory, LCAO 56 Hartree — Fock equations/theory, many-body perturbation 114—115 116 Hartree — Fock equations/theory, RHF 108—110 Hartree — Fock equations/theory, SCF 75 87 119 Hartree — Fock equations/theory, Slater’s Rules 54—56 Hartree — Fock equations/theory, solid state quantum mechanics 146—147 Hartree — Fock equations/theory, two-electron integrals 19 (see also “Fock” “Roothaan “UHF”) Hashed fingerprint 645—646 677 678 685 Heat bath, molecular dynamics 384 Heat capacity and computer simulation 308—309 348—349 Heitier — London model of hydrogen 124—125 Helium 36—38 39 Helium, hydrogen molecular ion ( 62—65 Helium, Slater determinant for 41 Helix 513—615 583 584 Hellmann — Feynman theorem 121 Helmholtz free energy 299—300 411 563—569 Helmholtz free energy, computer simulation 307 313—314 Hessian matrix 267—269 274—275 280 282—285 288 Heterovalent substituent 623 Heuristic searches and protein prediction 531—534 Hexane 449 462 463 672 Hexapeptide 482 HF 123 189 196 Hierarchical cluster analysis 494 534 High throughput screening (HTS) 641 High- 628—630 Hill potential 209 Histidine 169—170 510 525 556—557 HIV—1 protease 666 667 691—693 HMMs (Hidden Markov Models) 536—537 548 Hodgkin — Richards index 679 Hohnberg — Kohn theorem 128 Holonomic constraints 370 HOMO (highest occupied molecular orbit) 79 112 293—291 Hooke’s law 172—173 275 486 HP model 518—519 HTS (high throughput screening) 641 Huckel theory 99—102 Human genome project 512 548—549 Hunds rules 35 Hunter — Saunders approach 197—198 Hybrid Monte Carlo/molecular dynamics methods 452—453 Hydrodynamic vortex 377—378 Hydrogen 70 Hydrogen fluoride 620 Hydrogen, bonding 122—123 291—292 391 578 689 Hydrogen, bonding, bond order 83 Hydrogen, bonding, C-H bonds/interactions 98 167 180 211 233 236 362 Hydrogen, bonding, conformational analysis 490 504—505 Hydrogen, bonding, force fields 196 215—216 221 227—228 Hydrogen, bonding, new molecules 655 658 Hydrogen, bonding, O-H 98 620 Hydrogen, configuration interaction 112 Hydrogen, dissociation 109—110 Hydrogen, electron correlation 110—111 Hydrogen, energy minimisation methods 282—283 291—292 Hydrogen, fluoride 81—82 Hydrogen, Heitler — London model of 124—125 Hydrogen, molecule 41—46 Hydrogen, suppressed notation 644 Hydrophobic effect 515—517 518—519 669 Hysteresis 577 Iceberg model 516 ID3 algorithm 705 ILP (inductive logic programming) 705 Image charge computer simulation 340—341 Immunoglobulin 544 Immunosuppressant FK506 229—230 Importance sampling 410 Independent (random) samples 345 Indicator variable 696 INDO (intermediate neglect of differential overlap) 86 92—93 94—96 Inductive logic programming 705 Initial configuration, prior to simulation 315—316 Inorganic molecules, force fields for 234—236 Inorganic Structural Database 489 Inside-out ligand design 687—688 Integration, algorithms for molecular dynamics simulation 359—360 Integration, calculating properties by 412—414 Integration, thermodynamic 568—569 574 577 630—631 Intermediate neglect of differential overlap 86 92—93 94—96 Intermolecular processes and energy minimisation 278—279 Internal coordinates 2—4 257 Internet and World Wide Web 9—10 548 553 Interstitials 622—623 627 Intrinsic reaction coordinate 288—289 Inverse agonists 640 Inverse of matrix 15—16 Ionic solids, force fields for 238—240 Ionisation potentials 74—75 IRC (intrinsic reaction coordinate) 288—289 Iron, liquid 621—622 Isis system 645 Island model 481 Isodemic reactions 116 Isoleucine 511 525 556—557 Isomerism, subgraph 645 Isothermal-isobaric ensemble, definition 307 563 569 J-walking 533—535 Jahn — Teller effect 234 Jarvis — Patrick algorithm 496—497 683 JBW (jumping between wells) 435 Jellium 244 Jump frequency 627—628 Jumping between wells (JBW) 435 Kappa shape/kappa-alpha indices 672—674 Keys, pharmacophore 674—676 Kohn — Sham scheme/orbitals 128—129 131 132 134 135—137 156 157 616 Koopman’s theorem 74—75 Kronecker delta 30 396 kurtosis 680 Lag, ab initio molecular dynamics 618 Lagrange multiplier 18 52 127 191 371—372 Lags 453 Laguerre polynomials 31 55 Lambda dynamics 585—588 Langevin dipole method 601—603 Langevin equation 388—389 391 580 601—603 616 Langmuir — Blodgett films/layers 395 400—402 Large structures, reaction path for 289—292 Large systems, deriving charge models for 191—192 Latent variables 706 Lattices, models for proteins 518—520 Lattices, models of polymers 424—428 Lattices, solid state quantum mechanics 138—160 passim Lattices, statics and dynamics in energy minimisation 295—300 LCAO (linear combination of atomic orbitals) 41—42 56 100 241 LDA/LSDA <focal (spin) density approximation) 130—131 Leap-frog algorithm 356—357 Least-squares approach 230—231 Leave-one-out 701 Lee — Yang-Parr see “BLYP” Legendre polynomials 32 Length, units of 9 Lennard — Jones potential 253 Lennard — Jones potential, computer simulation 305 319 324 327 331—333 341—342 Lennard — Jones potential, force fields 167 207—310 212 214—216 225—226 237 Lennard — Jones potential, free energy calculations 579 586 613 Lennard — Jones potential, molecular dynamics simulation 361 368 402 Lennard — Jones potential, Monte Carlo simulation 428 439 441 448 450 LES (locally enhanced sampling) 575—576 Leucine 487 511 525 556—557 Levinthal paradox 550 Libraries, combinatorial 711—719 LIE (linear interaction energy) 588—589 Line search in one direction 262—263 Linear combination of atomic orbitals 41—42 56 100 241 Linear congruential method 418—419 Linear interaction energy 588—589 Linear potential, piecewise 665 Linear regression 666 698—699 702 Linear response 588—589 591 631—632 Linkage methods 493 Lipids 338 Lipids, simulation of 397—400 Liquid crystals 222—223 Literature 9 Lithium 111 238 323 626 Loadings 682 Local density approximation 130—131 Local spin DFT 129 135
Реклама



 )
) 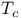 superconductor
superconductor 
 |
|
О проекте
|
|
О проекте