Авторизация
Поиск по указателям
Leach A.R. — Molecular Modelling Principles and Applications
Обсудите книгу на научном форуме Нашли опечатку?
Название: Molecular Modelling Principles and ApplicationsАвтор: Leach A.R. Аннотация: Preface to the Second Edition The impetus for this second edition is a desire to include some of the new techniques that have emerged in recent years and also extend the scope of the book to cover certain areas that were under-represented (even neglected) in the first edition. In this second volume there are three topics that fall into the first category (density functional theory, bioinformatics/protein structure analysis and chemoinformatics) and one main area in the second category (modelling of the solid state). In addition, of course, a new edition provides an opportunity to take a critical view of the text and to re-organise and update the material. Thus whilst much remains from the first edition, and this second book follows much the same path through the subject, readers familiar with the first edition will find some changes which I hope they will agree are for the better. As with the first edition we initially consider quantum mechanics, but this is now split into two chapters. Thus Chapter 2 provides an introduction to the ab initio and semi-empirical approaches together with some examples of the uses of quantum mechanics. Chapter 3 covers more advanced aspects of the ab initio approach, density functional theory and the particular problems of the solid state. Molecular mechanics is the subject of Chapter 4 and then in Chapter 5 we consider energy minimisation and other 'static' techniques. Chapters 6, 7 and 8 deal with the two main simulation methods (molecular dynamics and Monte Carlo). Chapter 9 is devoted to the conformational analysis of 'small' molecules but also includes some topics (e.g. cluster analysis, principal components analysis) that are widely used in informatics. In Chapter 10 the problems of protein structure prediction and protein folding are considered; this chapter also contains an introduction to some of the more widely used methods in bioinformatics. In Chapter 11 we draw upon material from the previous chapters in a discussion of free energy calculations, continuum solvent models, and methods for simulating chemical reactions and defects in solids. Finally, Chapter 12 is concerned with modelling and chemoinformatics techniques for discovering and designing new molecules, including database searching, docking, de novo design, quantitative structure-activity relationships and combinatorial library design. As in the first edition, the inexorable pace of change means that what is currently considered 'cutting edge' will soon become routine. The examples are thus chosen primarily because they illuminate the underlying theory rather than because they are the first application of a particular technique or are the most recent available. In a similar vein, it is impossible in a volume such as this to even attempt to cover everything and so there are undoubtedly areas which are under-represented. This is not intended to be a definitive historical account or a review of the current state-of-the-art. Thus, whilst I have tried to include many literature references it is possible that the invention of some technique may appear to be incorrectly attributed or a 'classic' application may be missing. A general guiding principle has been to focus on those techniques that are in widespread use rather than those which are the province of one particular research group. Despite these caveats I hope that the coverage is sufficient to provide a solid introduction to the main areas and also that those readers who are 'experts' will find something new to interest them.
Язык: Рубрика: Химия /Статус предметного указателя: Готов указатель с номерами страниц ed2k: ed2k stats Издание: 2-ndГод издания: 2001Количество страниц: 774Добавлена в каталог: 21.02.2007Операции: Положить на полку |
Скопировать ссылку для форума | Скопировать ID
Предметный указатель
Conformational analysis, clustering algorithms and pattern recognition 491—497 Conformational analysis, conformational search 457 662 Conformational analysis, crystal structures predicted 501—505 Conformational analysis, dimensionality of data set reduced 497—499 Conformational analysis, distance geometry 467—475 476 Conformational analysis, fitting, molecular 490—491 Conformational analysis, global energy minimum 458 479—483 Conformational analysis, model-building 464—465 Conformational analysis, poling 499—501 Conformational analysis, random 465—467 476 Conformational analysis, structural databases 482 489—490 493—494 499 Conformational analysis, systematic 458—464 476 505 Conformational analysis, variations on standard methods 477—479 Conformational changes in molecular dynamics simulation 392—393 Conformationally flexible docking 662—663 CONGEN program 542 Conjugate gradients 262 264—267 473 Conjugate peak refinement 290—291 Connection table 643 Consistent force field (CFF) 231 Constraints, chiral 473—474 Constraints, holonomic versus non-holomonic 370 Constraints, holonomic versus non-holomonic and restraints, difference between 369—370 Constraints, holonomic versus non-holomonic in molecular dynamics 368—374 Constraints, holonomic versus non-holomonic, simulation 368—374 Constraints, holonomic versus non-holomonic, subset selection 717 Constraints, holonomic versus non-holomonic, systematic search 649—651 Contact surface 7 Continuum models and solvation, free energy of 592—593 598—601 Contraction, basis set 69 Convergence sphere 186 coordinates 2—4 Coordinates, internal 2—4 257 Coordinates, intrinsic reaction 288—289 Coordinates, mass-weighted 274—275 Coordinates, scaled 438—439 (see also “Cartesian coordinates”) Corey — Pauling — Koltun (CPK) models 5—6 CORINA program 659 Correlation, BLYP 135 136 137 Correlation, coefficients 374 681 Correlation, electron 110—117 Correlation, exchange-correlation functional 129—134 Correlation, functions and molecular dynamics, simulation 374—380 Correlation, spectroscopy (COSY) 474—475 486 Cosine coefficient 676 685 COSMO (conductor-like screening model) 597 COSY (correlated spectroscopy) 474—475 486 Coulomb interaction/integral 598—599 Coulomb interaction/integral, advanced ab initio methods 122 127 128 132 133—134 146—147 Coulomb interaction/integral, computational quantum mechanics 30 42 45 49—53 58 60 85 100 Coulomb interaction/integral, force fields 184—185 187 194 202 238 Coulomb potential 167 244 338 341—342 Coulomb’s law 95 194 202 212 596 603—604 607 Counterpoise correction 121—122 Coupling parameter 567 Craig plot 681 Cross-correlation function 376 Crossover operator 480—481 Crystal momentum 148 Crystal structures, predicted 501—505 CSD (Cambridge Structural Database), conformational analysis 482 489 493—494 499 CSD (Cambridge Structural Database), new molecules 659 691 693 Cu-Zn superoxide dismutase 607 Cut-offs in computer simulation 324—327 Cut-offs in computer simulation, group-based 327—330 Cut-offs in computer simulation, problems with 330—334 Cyclic urea, HIV protease inhibitor 691—693 Cyclobutane 176—177 Cyclobutanone 176 Cyclobutene 117 Cycloheptadecane 476 Cyclohexane 286 463 465 497 597 644 Cyclopropene 117 Cyclosporin 196—197 391 Cysteine 511 525 556—557 Cytosine 84 227 D-Glucose 575 D-optimal design 697—698 databases 489 537 539 Databases, 3D 659—661 679 “Structural Davidon — Fletcher — Powell method 269—270 de Broglie thermal wavelength 411 440—441 de novo ligand design 687—694 Degrees of freedom 699—700 Delocalised 233—234 DelPhi program 604—605 Density, charge density matrix 58—59 Density, electron 77—79 80—2 160 Density, functional theory 126—137 156 619 Density, functional theory of levels 154 Density, spin 129—131 Density, spin of states and Fermi surface 153—155 Depth-first search 462 663 Derivative, energy, calculating 120—121 Derivative, energy, function 225—226 Derivative, energy, minimisation 257—258 261—262 268—269 Descriptors and new molecules 668—679 Determinant of matrix 13—14 DFP (Davidon — Fletcher — Powell) method 269—270 DFT (density functional theory) 126—137 156 619 DHFR (dihydrofolate reductase) 278—279 320 460 Diagonalisation of matrix 16 Diatomic overlap see “MNDO” Dick — Overhauser shell model 239 dielectric constant 297 Dielectric models 202—204 Diels — Alder reaction 294—295 615 716—717 Differences, free energy 564—574 Differences, free energy, applications of methods 569—574 Differences, free energy, formula for 568 630—631 Differences, free energy, methods for calculating 564—569 Differential overlap, neglect of 86 89—96 Differential overlap, zero (ZDU) 88—89 91 Dihedral angle, definition 4 Dihydrofolate reductase 278—279 320 460 DIIS (direct inversion of iterative subspace) 118 Dimensionality reduction 497—499 Dimethyl formamide 613—614 Dimethyl thioether 676—677 Dipole 75—77 Dipole correlation time 378—378 Dipole, force fields 181 182—185 189 199—201 219 246 Dipole, models of solvation, free energy of 593—595 601—603 Dipole, moment, net 378—379 Dipole, triple 213—215 239 Direct inversion of iterative subspace 118 Direct SCF method 118—120 Director 396 Discriminant analysis and QSAR 703—705 Dispersion curve 298—299 Dispersive interactions 204—206 Displacements 322—323 624 Dissimilarity-based methods 683—685 Dissipative particle dynamics 402—404 Distance, bounds 468 Distance, bounds, geometry 467—475 476 651—653 663 Distance, bounds, Hamming (city block) 492 676—678 Distance, bounds, map 651 Distance, bounds, matrix 652 Distance, bounds, Soergel 676—678 Distance-dependent dielectric 203 Distributed multipole analysis 195—197 Diverse sets of compounds, selecting 680—687 DMA (distributed multipole analysis) 195—197 DMF (dimethyl formamide) 613—614 DNA 197 227 452 489 604 DNA, computer simulation 319 338—339 DNA, Human Genome Project 512 548—549 DNA, inhibitor 270—271 DNA, new molecules 662 DNA, proteins 509 512 549 DOCK program/algorithm 662—663 665 667 docking 661—668 689 Domain 515 Double dynamic programming 537—539 Double zeta basis sets 70 Double-wide sampling 567—568 DPD (dissipative article dynamics) 402—404 Dreiding models 5—6 Drude molecules 205—206 Drude molecules, interaction between 246—247 Drugs, new see “New molecules” Dual topology 578 Dummy atoms, in Z-matrix 271—272 Dunning basis sets 73 Dynamic/ dynamics 353—409 Dynamically modified windows 578 Dynamically modified windows and statics in energy minimisation 295—300 (see also “Molecular dynamics”) Dynamically modified windows, programming and protein prediction 526—529 EA (evolutionary algorithms) 479—483 Edges of search trees 461 Effective medium theory 243—241 Effective pair potentials 214—215 Eigenvalues and eigenvectors 15—16 114 Eigenvalues and eigenvectors, conformational analysis 469 471 479 498 Eigenvalues and eigenvectors, energy minimisation methods 272 282—285 Einstein relationships 381 627—628 Eisenberg’s 3D profiles 543—544 Elastic constants 240 296—297 Electric multrpoles, calculation of 75—77 Electron, affinity (EA) 194 Electron, correlation 110—117 Electron, density 77—79 80—82 160 Electron, gas theory 240 Electron, integrals, one- and two- 50—51 Electron, nearly free-electron approximation 147—153 Electron, polyelectronic atoms and molecules 34—41 Electron, spin 34—35 38 Electronegativity 192—193 Electrostatic interactions, force fields 166 181—204 221 237 Electrostatic interactions, free energy calculations 566—567 576 580 588 613 Electrostatic interactions, potentials 83—85 188 189—191 Electrostatic interactions, solvation free energy calculations 593—608 Electrotopological state index 674 Embedded-atom model 241 243—244 Embedding 469 Empirical bond-order potential see “Tersoff” Endothiapepsin 589—591 Energy minimisation methods 253—302 623 Energy minimisation methods, applications of 273—279 Energy minimisation methods, choice of 270—273 Energy minimisation methods, derivative 257—258 261—262 268—269 Energy minimisation methods, first-order 262—267 Energy minimisation methods, Newton — Raphson 267—268 270 288 Energy minimisation methods, non-derivative 258—261 Energy minimisation methods, quasi-Newton 268—269 Energy minimisation methods, solid-state systems 295—300 Energy minimisation methods, statement of problem 255—257 Energy minimisation methods, transition structures and reaction pathways 279—295 Energy of closed-shell system 51 Energy of general polyelectronic system 46—50 Energy, calculation from wavefunction 41—46 Energy, component analysis 122—124 Energy, computer simulation 308 348—349 Energy, conservation in molecular dynamics simulation 359 405—406 Energy, derivatives, calculating 120—121 Energy, force field 240 Energy, function, derivatives of 225—226 Energy, global minimum 253 458 479—483 551—552 Energy, Koopman’s theorem and iorusation potentials 74—75 Energy, lower-energy regions 564 Energy, minimum, global 253 458 479—483 551—552 Energy, potential 4—5 238 253 Energy, strain 226—7 627 Energy, surface (hypersurface) 4—5 253 475 Energy, units of 9 (see also “Derivative” “Energy “Free “Quantum Enol boroate/aldehyde reaction 610—611 Ensemble, averages 303—305 Ensemble, distance geometry 651—653 Ensemble, molecular dynamics 653 Ensemble, Monte Carlo simulation 438—442 450—451 Enthalpy 159 574 entropy 574 Enumeration of libraries 715—717 Equilibration monitoring and computer simulation 321—323 Equilibria phases in computer simulation 315 450—451 Ergodicity 304 313 Ergodicity, quasi ergodicity 433—438 Error, estimating in a simulation 343—347 ESS (explained sum of squares) 699—700 Ethane, carbon-carbon bond 4—5 Ethane, force fields 174 Ethane, Monte Carlo simulation 441—442 Ethane, SMILES notation 644 Ethane, thiol 564—569 Ethane, torsion angles 286—287 Ethane, Z-matrix 2—3 9 Ethanol 564—569 611—612 Ethene 83 236 293—295 ethylene 621 Ethyne 83 Euclidean distance measure 492 Euler angles 421—422 Even-tempered basis set 71—72 Evolutionary algorithms 479—483 Evolutionary design 694 Evolutionary planning (EP) and strategies (ES) 479—480 482 Ewald summation 238 334—339 342 402 625—626 Exchange, correlation functional 129—134 Exchange, forces see “Repulsive forces” Exchange, gradient 135 136 137 Exchange, integral 50 52—53 58 60 Exchange, interaction 46 Exclusion spheres 658—659 Exons 512 Explained sum of squares 699—700 Extended Hiickel theory (EHT) 101—102 Extended system method 384 Extreme value distribution 532 Fabric softeners 401—402 Face-centred cubic lattice 139 316 Factor analysis 681—682 686 Factors (variables) 697 Family (proteins) 539 Fast Fourier Transform 24 338—339 342 Fast multipole method 341—343 364 FASTA 524 531 FDPB (finite difference Poisson — Boltzmann method) 604—608 Fermi surface and energy 153—155 Ferrocene 234 FFT (fast Fourier transform) 24 338—339 342 Fick’s laws 380—381 Finite difference methods 355—358 604—608 Finnis — Sinclair potential 241—245 passim First principles method for predicting proteins 517—522 First-order energy minimisation 262—267 Fitting, molecular 490—491 Flexible fitting 491 Flexible molecules 423 582—585 FlexX program 667 Fock matrix 100 Fock matrix, Hartree — Fock equations 57—59 61 63—64 Fock matrix, open-shell systems 108—109 Fock matrix, semi-empirical methods 89—90 94 95 96 Fock matrix, solid state quantum mechanics 146 Fock operator 53 57 114 Focusing 606 Folding see “Under proteins” Force field models, empirical 165—252 610 Force field models, empirical, 233—234
Реклама



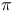 -systems, force fields for
-systems, force fields for  |
|
О проекте
|
|
О проекте