Авторизация
Поиск по указателям
Leach A.R. — Molecular Modelling Principles and Applications
Обсудите книгу на научном форуме Нашли опечатку?
Название: Molecular Modelling Principles and ApplicationsАвтор: Leach A.R. Аннотация: Preface to the Second Edition The impetus for this second edition is a desire to include some of the new techniques that have emerged in recent years and also extend the scope of the book to cover certain areas that were under-represented (even neglected) in the first edition. In this second volume there are three topics that fall into the first category (density functional theory, bioinformatics/protein structure analysis and chemoinformatics) and one main area in the second category (modelling of the solid state). In addition, of course, a new edition provides an opportunity to take a critical view of the text and to re-organise and update the material. Thus whilst much remains from the first edition, and this second book follows much the same path through the subject, readers familiar with the first edition will find some changes which I hope they will agree are for the better. As with the first edition we initially consider quantum mechanics, but this is now split into two chapters. Thus Chapter 2 provides an introduction to the ab initio and semi-empirical approaches together with some examples of the uses of quantum mechanics. Chapter 3 covers more advanced aspects of the ab initio approach, density functional theory and the particular problems of the solid state. Molecular mechanics is the subject of Chapter 4 and then in Chapter 5 we consider energy minimisation and other 'static' techniques. Chapters 6, 7 and 8 deal with the two main simulation methods (molecular dynamics and Monte Carlo). Chapter 9 is devoted to the conformational analysis of 'small' molecules but also includes some topics (e.g. cluster analysis, principal components analysis) that are widely used in informatics. In Chapter 10 the problems of protein structure prediction and protein folding are considered; this chapter also contains an introduction to some of the more widely used methods in bioinformatics. In Chapter 11 we draw upon material from the previous chapters in a discussion of free energy calculations, continuum solvent models, and methods for simulating chemical reactions and defects in solids. Finally, Chapter 12 is concerned with modelling and chemoinformatics techniques for discovering and designing new molecules, including database searching, docking, de novo design, quantitative structure-activity relationships and combinatorial library design. As in the first edition, the inexorable pace of change means that what is currently considered 'cutting edge' will soon become routine. The examples are thus chosen primarily because they illuminate the underlying theory rather than because they are the first application of a particular technique or are the most recent available. In a similar vein, it is impossible in a volume such as this to even attempt to cover everything and so there are undoubtedly areas which are under-represented. This is not intended to be a definitive historical account or a review of the current state-of-the-art. Thus, whilst I have tried to include many literature references it is possible that the invention of some technique may appear to be incorrectly attributed or a 'classic' application may be missing. A general guiding principle has been to focus on those techniques that are in widespread use rather than those which are the province of one particular research group. Despite these caveats I hope that the coverage is sufficient to provide a solid introduction to the main areas and also that those readers who are 'experts' will find something new to interest them.
Язык: Рубрика: Химия /Статус предметного указателя: Готов указатель с номерами страниц ed2k: ed2k stats Издание: 2-ndГод издания: 2001Количество страниц: 774Добавлена в каталог: 21.02.2007Операции: Положить на полку |
Скопировать ссылку для форума | Скопировать ID
Предметный указатель
701 699 612—614 280—282 513—515 513—514 513 197—199 126 233—234 1, 2-dichloroethane 387—388 1, 3, 5-trifluorobenzene 198—199 1-octanol and water, partition between 668—669 2-methyl propane 644 2D substructure searching 642—647 4-acetamido benzoic acid 661 4-methyl-2 oxetanone 137 ab initio defined 65 ab initio molecular dynamics 616—622 ab initio potentials for water 216—218 ab initio quantum mechanics, calculating properties using 74—86 (see also “Advanced ab initio methods”) Absolute free energies 573—574 Accessible surface 7 ACE (angiotensin converting enzyme) 649—651 Acetaldehyde 180 578 Acetamide 573 Acetic acid 504—505 573 643 Acetic acid, SMILES notation 644 645 Acetonitrile 597 Acetylcholine 678 Acronyms and abbreviations 104—105 553—554 Adenine 227 Adiabatic mapping 286 Adjacency matrix 647 Adjoint matrix 15 Adsorption processes, Monte Carlo simulations of 441—442 Advanced ab initio methods 108—164 Advanced ab initio methods, density functional theory 126—137 Advanced ab initio methods, electron correlation 110—117 Advanced ab initio methods, energy component analysis 122—124 Advanced ab initio methods, open-shell systems 108—110 Advanced ab initio methods, practical considerations 117—122 Advanced ab initio methods, solid state quantum mechanics 138—160 Advanced ab initio methods, valence bond theories 124—126 Agglomerative cluster analysis methods 493—494 Agonists 640 AINT function 350 Alanines 169 459 511 525 542 546 556—557 Alanines, energy minimisation methods 277 280 286 Alanines, free energy calculations 583—584 Aldehyde 610—611 Aldol reactions 610—612 Aliovalent substitution 623 Alkaline earth oxides 147 Alkanes 449—450 AMBER force field 169—170 175—176 191 211 230—232 AMI 86 97—98 102—103 230 Amino acids 511 549 602 Amino acids, computer simulation 329—330 Amino acids, conformational analysis 459 487 Amino acids, energy minimisation methods 277 280 286 Amino acids, force fields 169—170 221 Amino acids, free energy calculations 572 583—584 Amino acids, motifs 522 Amino acids, PAM matrices 524—526 531 556—557 Amino acids, torsion angles 515 (see also “Peptides; proteins”) Amino acids, ’threading’ 546 Aminothlazoles 716 AMP AC program 8 99 Amphiphiles, molecular dynamics simulation of 394—404 Angiotension converting enzyme 649—651 Angle bending 166 173 Annealing, simulated 483—489 504—505 519 691 Annotations 513 Antagonists 640 Antisymmetry principle 35 Arbitrary step energy minimisation 264 Argand diagram 17 Arginme 329—330 510 525 556—557 Argon 253 323 Argon, force fields 205 214 Argon, J-walking 434—435 Argon, time-steps 361—362 Argon, velocity autocorrelation 377 Arithmetic mean 20 Aromatic systems and charge schemes 197—199 Asparagine 330 510 525 546 556—557 Aspartic acid 510 525 556—557 Atoms in molecules theory 80—81 Atoms/atomic, charges 157—159 181 192—195 Atoms/atomic, marker 329—330 Atoms/atomic, one-electron 30—34 Atoms/atomic, orbitals 41—42 56 100 241 Atoms/atomic, polyelectronic 34—41 Atoms/atomic, type 169 Atoms/atomic, units 29 Aufbau principle 35 Austin Model I (AMI) 86 97—98 102—103 Autocorrelation function 376—378 Automated protein modelling 548—549 Autoscaling 681 Availability 300 Axilrod — Teller term (triple-dipole) 213—215 239 Azimuthal quantum number 31 B3LYP density function 615 Backtracking 462 Backward sampling 567 band theory 141—142 Band theory, orbital-based approach 142—146 Barker — Fisher — Watts potential 214 Basic Local Alignment Search Tool see “BLAST” Basic Local Alignment Search Tool, basis sets/functions 56 85 123 Basic Local Alignment Search Tool, computational quantum mechanics 65—74 Basic Local Alignment Search Tool, Gaussian functions 65—73 passim 120 137 195 Basic Local Alignment Search Tool, superposition error 121—122 (see also “STOs”) BCUT method 686—687 Bead model of polymers 428 Becke see “BLYP” Beeman’s algorithm 357 Bending 166 173 176—178 Benperidol 675 Benzamidine 586—588 benzene 123 Benzene, force fields 170 174 178 186 197—198 Benzene, Huckel theory 99 100 Benzene, ring 81 170 178 Benzene, SMILES notation 644 Benzene, spin-coupled valence bond theory 126 Benzyl bromide 670 Beryllium 40 113 BFGS(Broyden-Fletcher-Goldfarb-Shanno) method 269—270 Bilinear model 702 Binding site 662 689—691 Binomial expansion 11 Bioinformatics 513 (see also “Amino acids” “DNA” “Proteins”) Bioisosteres 648 Biotin 576 641 bitstring 645—647 BLAST (Basic Local Alignment Search Tool) 521 524 531—534 548 Bloch’s theorem/function 142—145 146 148 161 Block-diagonal Newton — Raphson minimisation 268 BLOSUM matrices 526 BLYP (Becke gradient-exchange correction and Lee — Yang — Parr correlation functional) 135 136 137 Bohr radius 31 Boltzmann distribution 192 214 274 483 611 Boltzmann distribution, computer simulation 306—307 347 Boltzmann distribution, conformational analysis 457 Boltzmann distribution, Monte Carlo simulation 415—416 433 435—436 445—446 Boltzmann factor 306—307 413 Boltzmann weighted average 576 581 Bond fluctuation model 424—425 Bond/bonding 644 Bond/bonding, inorganic molecules 234—235 Bond/bonding, lack of see “Non-bonded interactions” Bond/bonding, orders 81—83 Bond/bonding, stretching 166 170—173 Bond/bonding, valence 124—126 (see also “Under carbon” “Hydrogen”) Born equation/model 238 593—594 598—601 609 Born — Oppenheimer approximation 4 35—36 50 Boundary, computer simulation 317—321 Boundary, element method 598 Bravais lattices 138—139 Brillouin’s theorem 112—113 115 Brilloum zone 140—141 145—146 150—151 157—158 298—299 Bromine 571 Broyden — Fletcher — Goldfarb — Shanno method 269—270 BSSE (basis set superposition error) 121—122 Buckingham potential 209 238 Build-up approach 517 Butadiene 233 294—295 Butane 85—86 186 449—450 582 Butanone 611—612 Cage structure of liquids 377 Calcium 589 626 Calix[4]arene 291—292 Cambridge structural database see “CSD” canonical ensemble 563 569 Canonical genetic algorithms 479—480 Canonical representation 644 Canonical structures 541 Captopril 649 Car — Parrinello scheme 610 617—619 Carbo index 678—679 Carbon dioxide 181 316 616—617 Carbon, bonds 4—5 98 362 612 652 Carbon, bonds, energy minimisation methods 253 280—281 Carbon, bonds, force fields 167 180 211 233 236 Carbon, five-carbon fragment 472 Carbon, force fields 167 180 211 233 236 244 Carbon, valence electron density 160 Carbonic anhydrase 616 Carboxylic acid 660 Cartesian coordinates 2—4 Cartesian coordinates, conformational analysis 466—468 Cartesian coordinates, energy minimisation methods 255 257—258 275 290 Cartesian coordinates, molecular dynamics simulation 370 372 379 393 Cartesian coordinates, Monte Carlo simulation 417 420—421 423 Cartesian coordinates, vectors 11—12 CASP3 548 CASSCF (complete active-space SCF) 113 295 CAVEAT program 689 CBMC (configurational bias Monte Carlo) simulation 443—50 Cell, cubic 315—319 Cell, index method 326 Cell, multipole method 341—343 Cell, unit 138 Cell, Wigner — Seitz 140 350 Central dogma 509 512 Central multipole expansion 181—187 CFF (consistent force field) 231 Chain amphiphiles and molecular dynamics 394—404 Charge 187 Charge, atomic 157—159 181 191—195 Charge, density matrix 58—59 Charge, image simulation 340—341 Charge, oscillating 201—202 Charge, schemes 197—199 CHELP procedure 191 Chemical potential calculation in Monte Carlo simulation 442—443 Chemical reactions 610—622 Chemical reactions, molecular dynamics 616—622 Chemical reactions, potential of mean force of 612—614 Chemical reactions, quantum and molecular mechanics combined 614—616 Chemical reactions, simulation empirically 610—612 Chemokine 475 Chi molecular connectivity indices 672 Chi-squared test 344—345 Chiral constraints 473—474 Chlorides 238 612—613 614 676—677 Chlorine 181 231 280—281 571 612 620—621 Chloroform 227 573 Chlorpromazine 678 Chymosin 545 Chymotrypsin 522—523 CI (configuration interaction) 111—113 120 CID (configuration interaction doubles) 112 113 CISD (configuration interaction singles and doubles) 112 113 City block see “Hamming” Clausius — Mosotti relationship 238—239 Clausius, virial theorem of 309 Clique detection 653—656 CLOGP program 669—670 Closed-shell systems 51 56—9 86—88 109 Cluster analysis 494 534 682—683 Clustering algorithms 491—497 CMR program 671 CNDO (complete neglect of differential overlap) 86 89—92 93 94 95 Coefficients 148—149 374 Coefficients, new molecules 676—678 680—681 685 Coefficients, partition 572—573 668—671 Cofactor 14—15 Combination generator 420 453—454 Combinatorial explosion 460—461 Combinatorial libraries 711—719 CoMFA (comparative molecular field analysis) 679 708—711 Comparative modelling of proteins 539—545 Comparative molecular field analysis 679 708—711 Complete active-space SCF 113 295 Complete neglect of differential overlap 86 89—92 93 94 95 Complete-linkage (furthest-neighbour) cluster algorithm 493—494 complex numbers 16—18 Computational quantum mechanics 26—107 Computational quantum mechanics, acronyms used in 104—105 Computational quantum mechanics, approximate orbital theories 86 Computational quantum mechanics, atomic units 29 Computational quantum mechanics, basis sets 65—74 Computational quantum mechanics, calculating properties 74—86 Computational quantum mechanics, Huckel theory 99—102 Computational quantum mechanics, one-electron atoms 30—34 Computational quantum mechanics, operators 28—29 Computational quantum mechanics, polyelectroruc atoms and molecules 34—41 Computational quantum mechanics, semi-empirical methods 65 86—99 102—103 “Hartree Computer simulation 303—352 Computer simulation, boundaries 317—321 Computer simulation, equilibration, monitoring 321—323 Computer simulation, free energy calculation difficulties 563—564 Computer simulation, long-range forces 334—343 Computer simulation, molecular dynamics 305—306 307 Computer simulation, phase space 312—315 Computer simulation, practical aspects 315—316 Computer simulation, real gas contribution to virial 309 349—350 Computer simulation, results and errors 343—347 Computer simulation, statistical mechanics 347—348 Computer simulation, thermodynamic properties, simple 307—312 Computer simulation, time and ensemble averages 303—305 Computer simulation, translating particle back into control box 350 Computer simulation, truncating potential and minimum image convention 324—334 (see also “Molecular dynamics simulation Monte Computers, hardware 8—9 Computers, Internet and World Wide Web 9—10 548 553 Computers, software 8—9 99 concepts 1—25 (see also “Coordinates” “Mathematical CONCORD program 659 Conditionally convergent series 336 Conduction band 142 Conductor-like screening model 597 Configuration interaction see “CI” “CID” “CISD” Configurational bias see “CBMC” Conformational analysis 457—508 Conformational analysis and NMR/x-ray crystallography 468 474—475 483—489 Conformational analysis, choice of method 476—477
Реклама



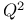 (cross-validated R2)
(cross-validated R2) 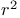
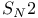 reaction, potential of mean force
reaction, potential of mean force 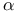 -Helix
-Helix  -strand structures
-strand structures  systems
systems  |
|
О проекте
|
|
О проекте